Hi, I’m Pelayo González de Lena
I am a computational biologist and bioinformatician working on
histone post-translational modifications (hPTMs) in plants, with a special
focus on H3K79 methylation and acetylation in Arabidopsis thaliana.
I combine quantitative mass spectrometry, reproducible computational pipelines, and
long-read transcriptomics to understand how chromatin states change across development and stress.
PhD Thesis: H3K79 in Arabidopsis
Currently finishing my PhD at the University of Oviedo (Department of Organisms
and Systems Biology), funded by an FPI fellowship (PRE2019-091395). My thesis
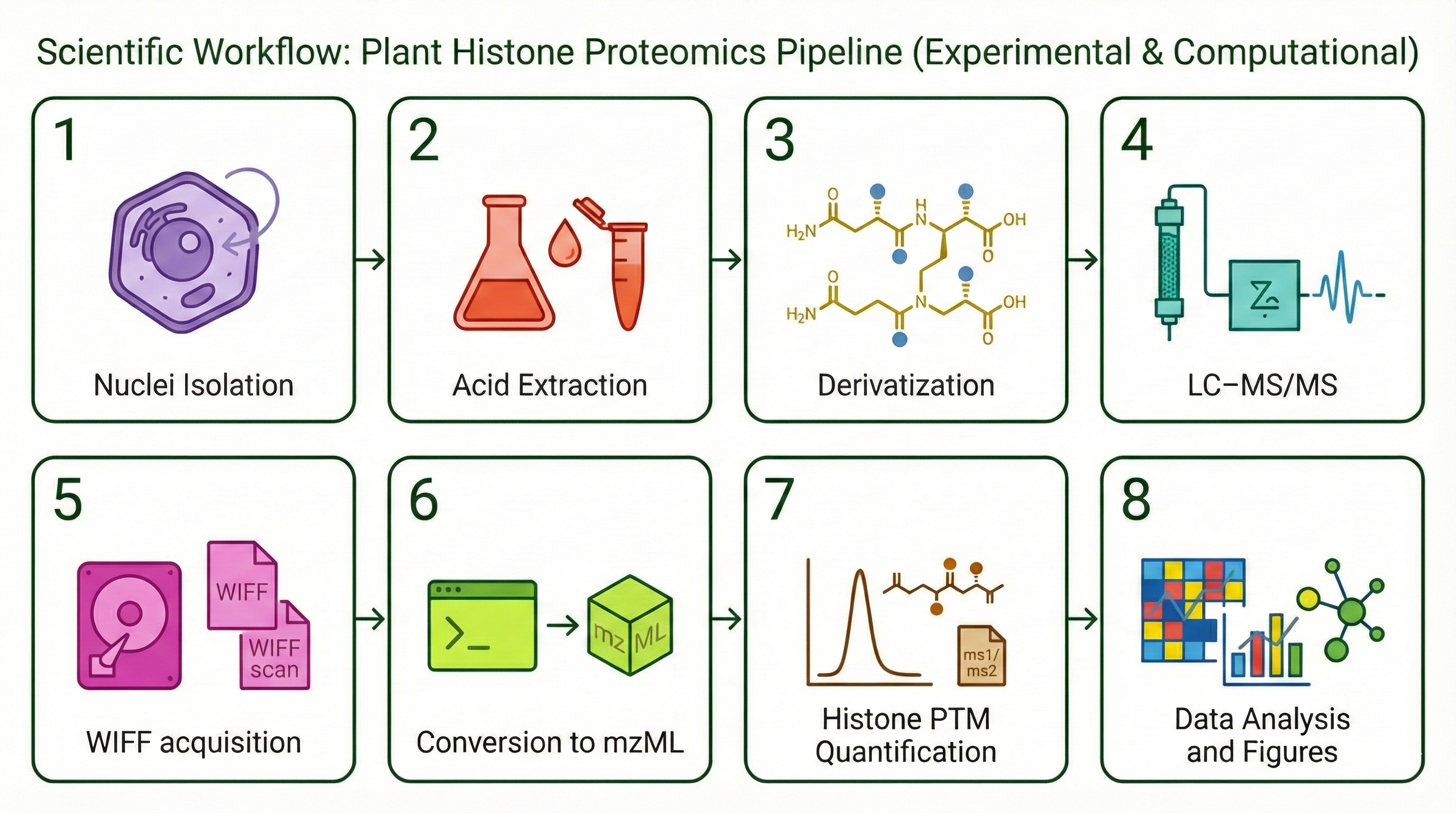
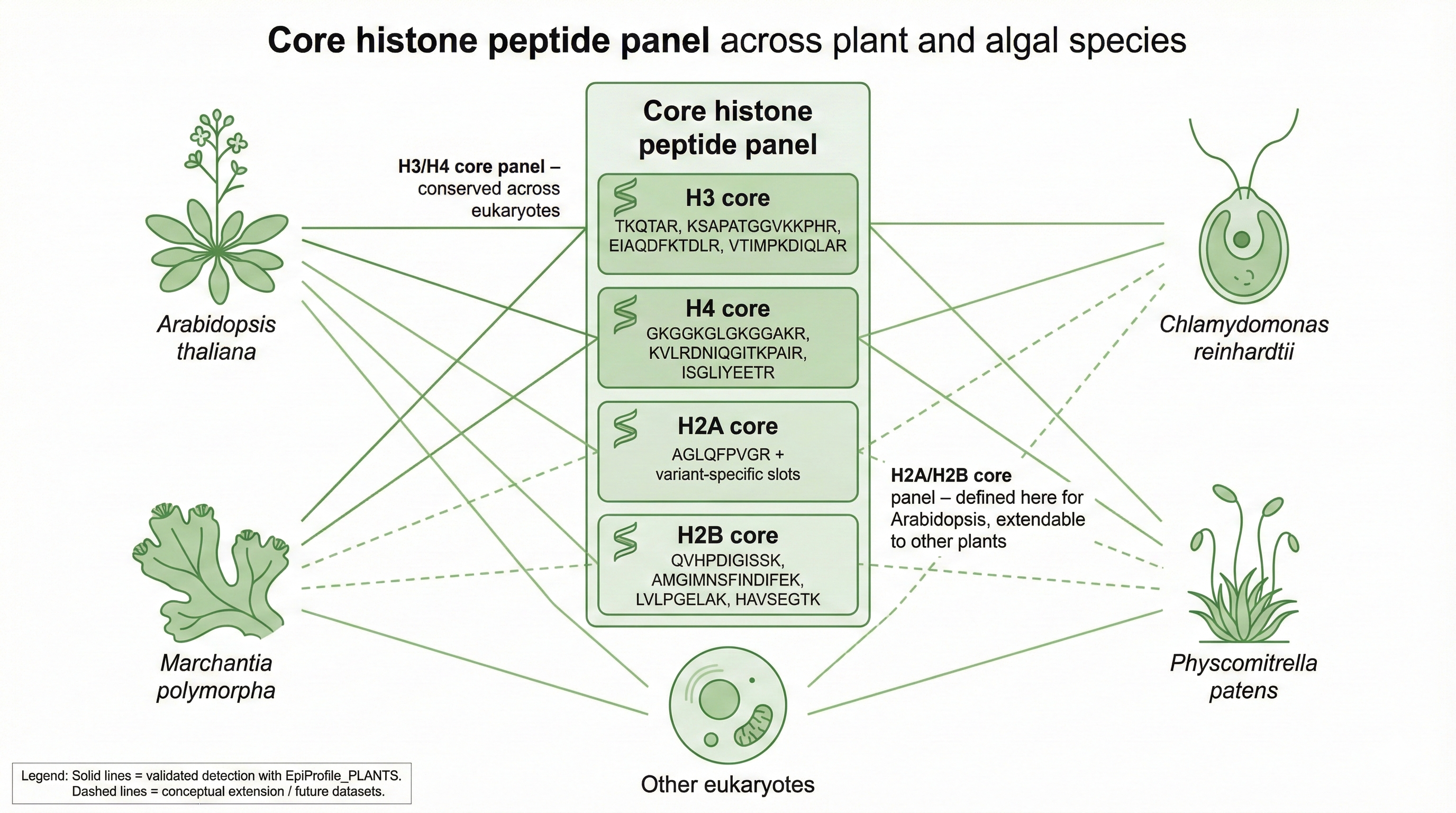
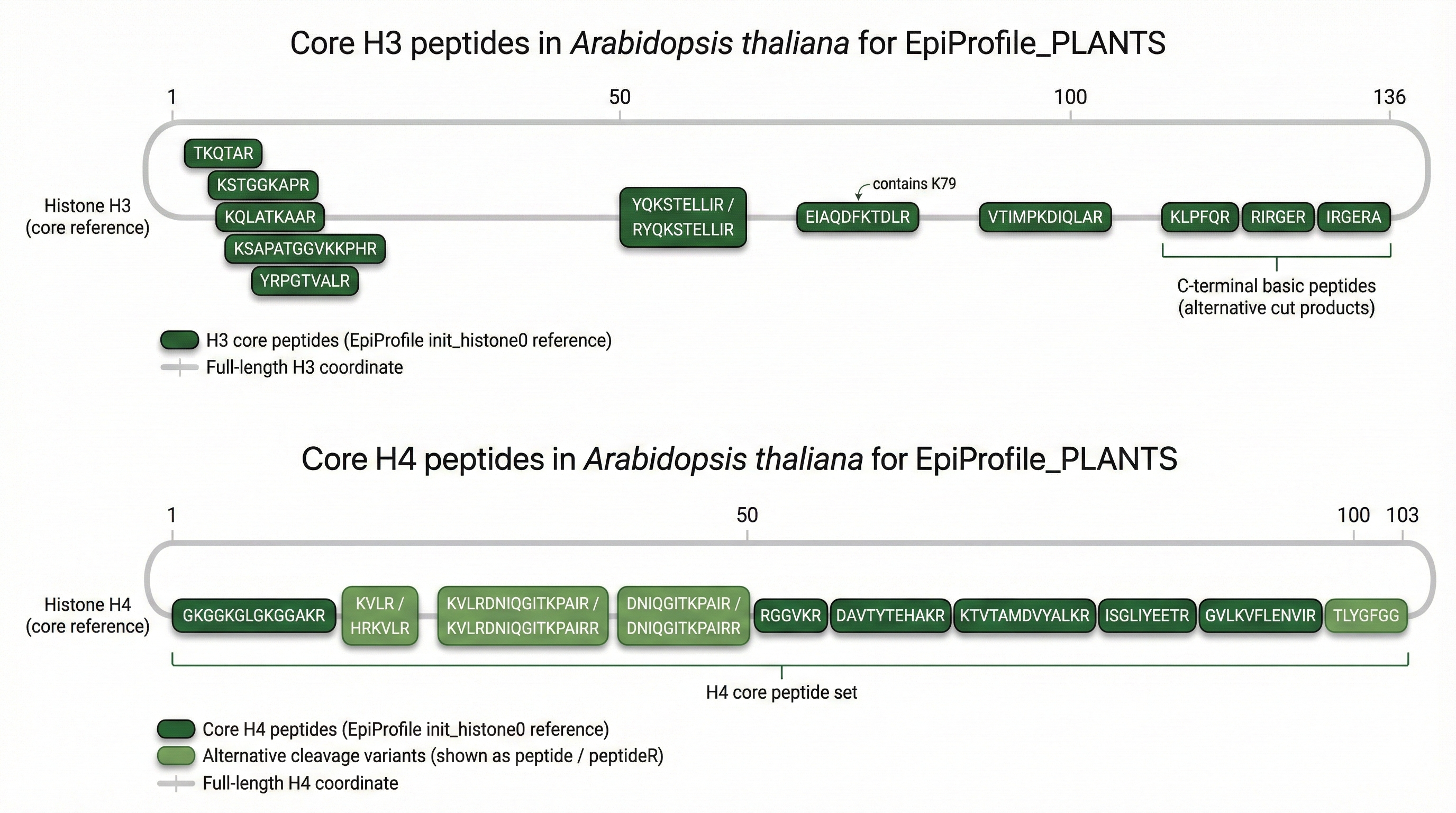
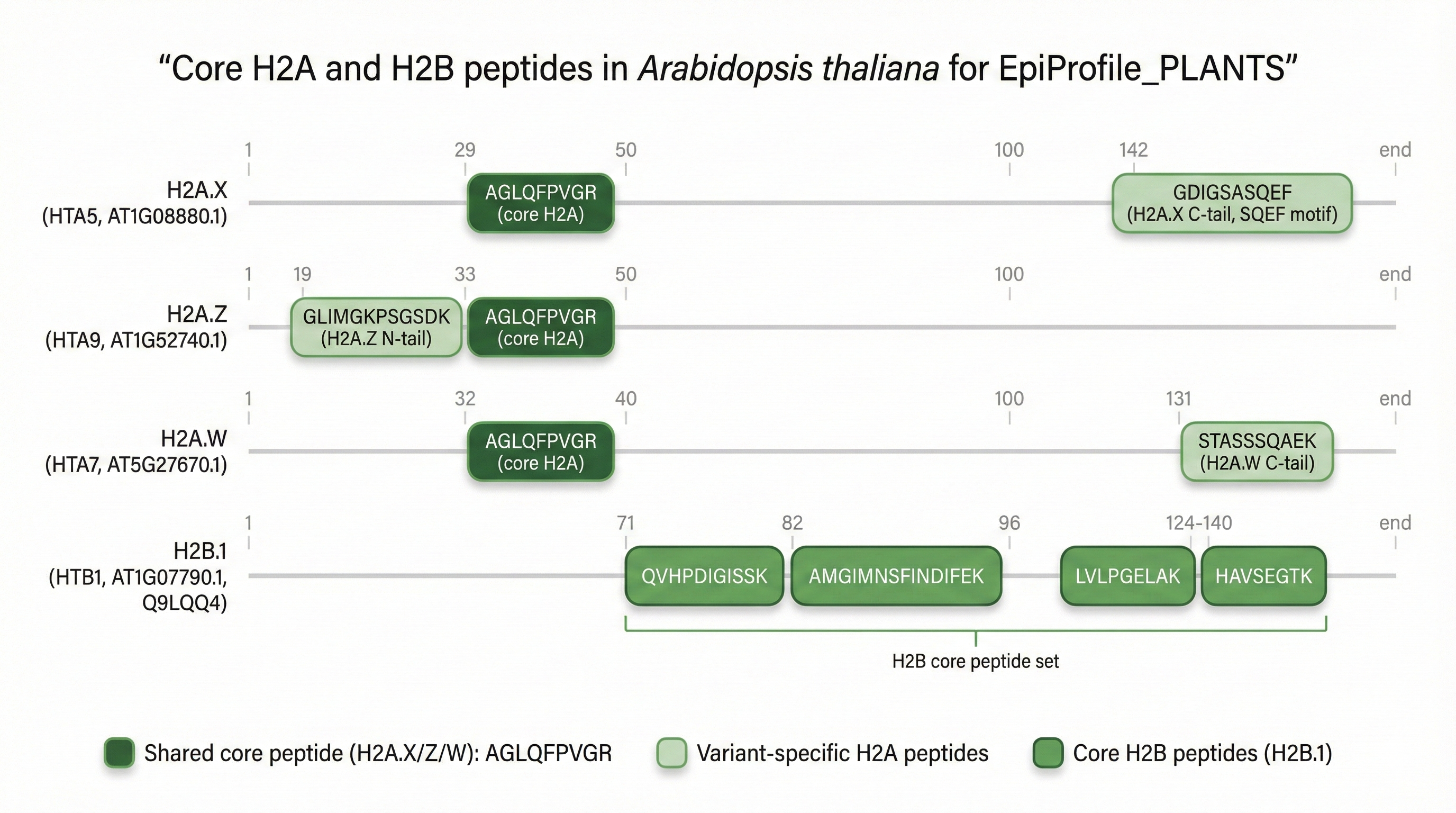
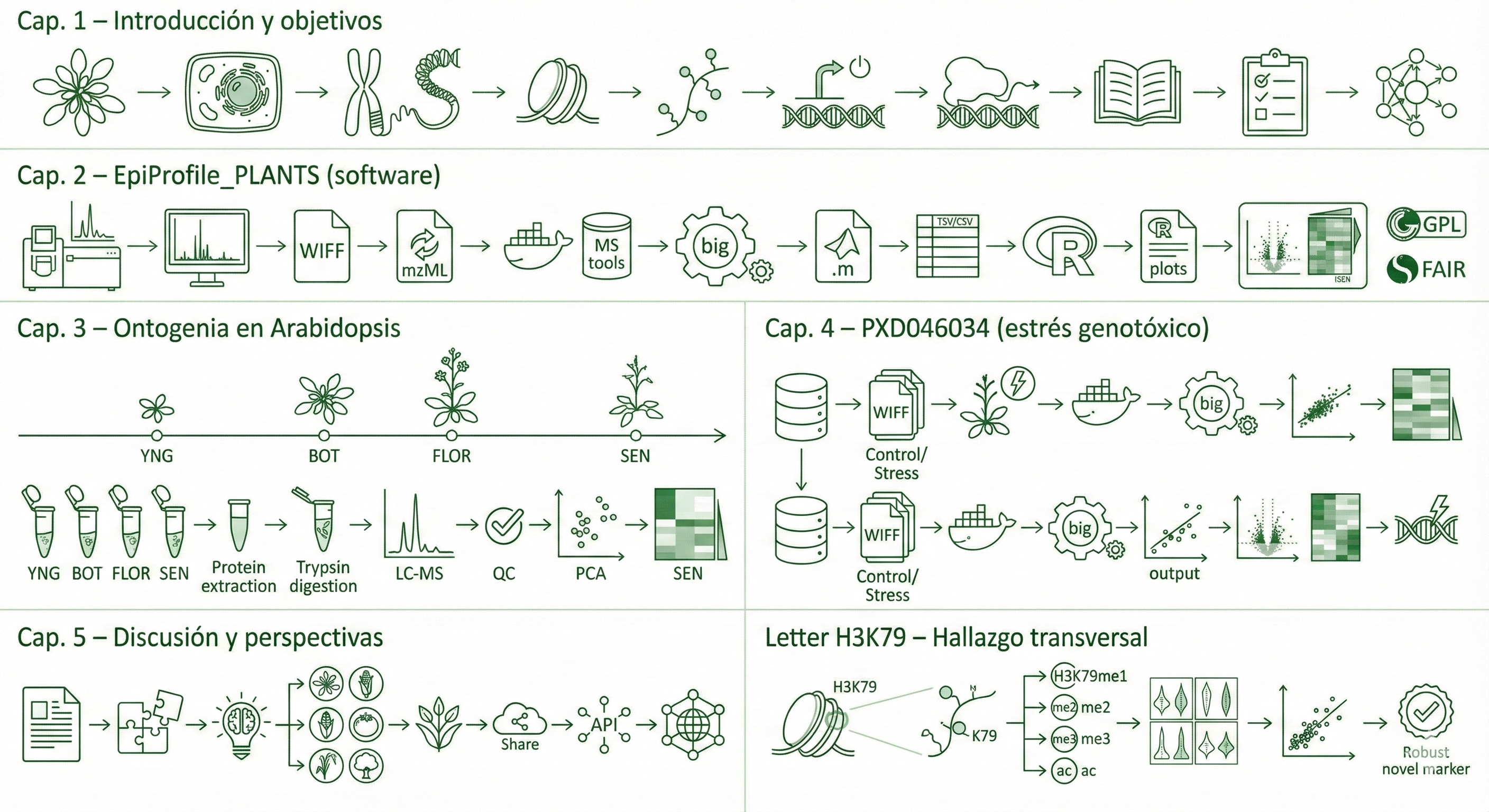
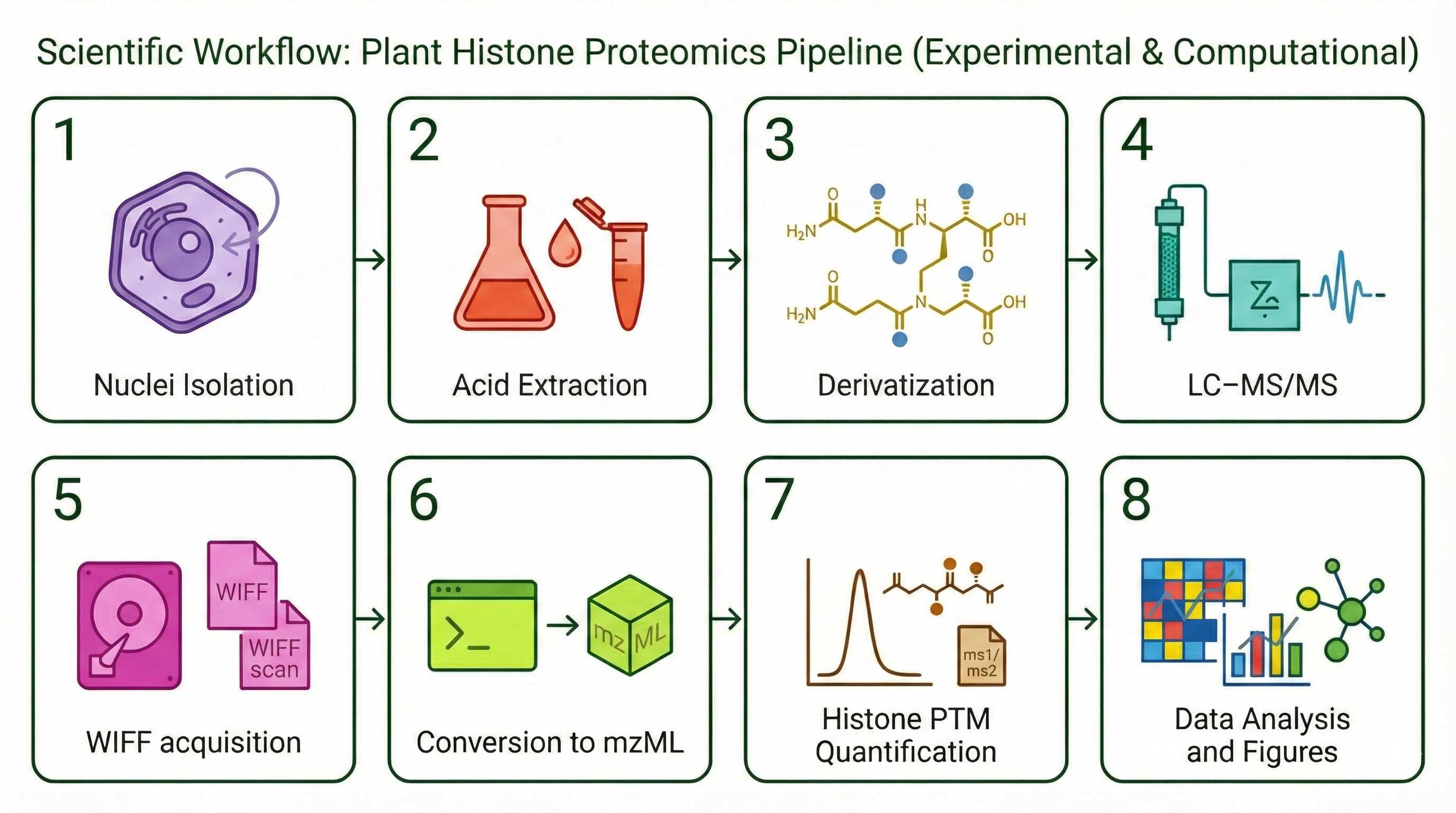
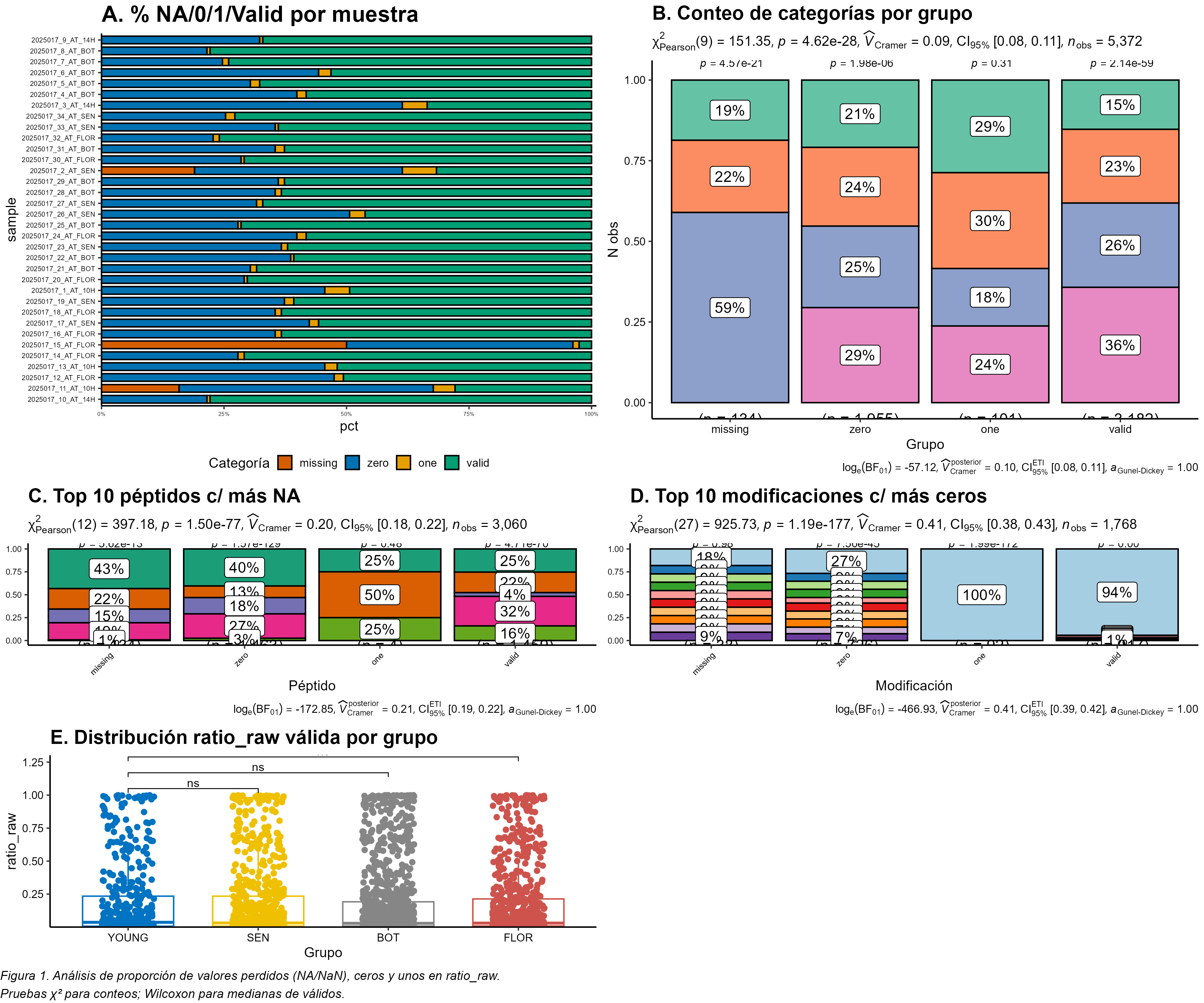
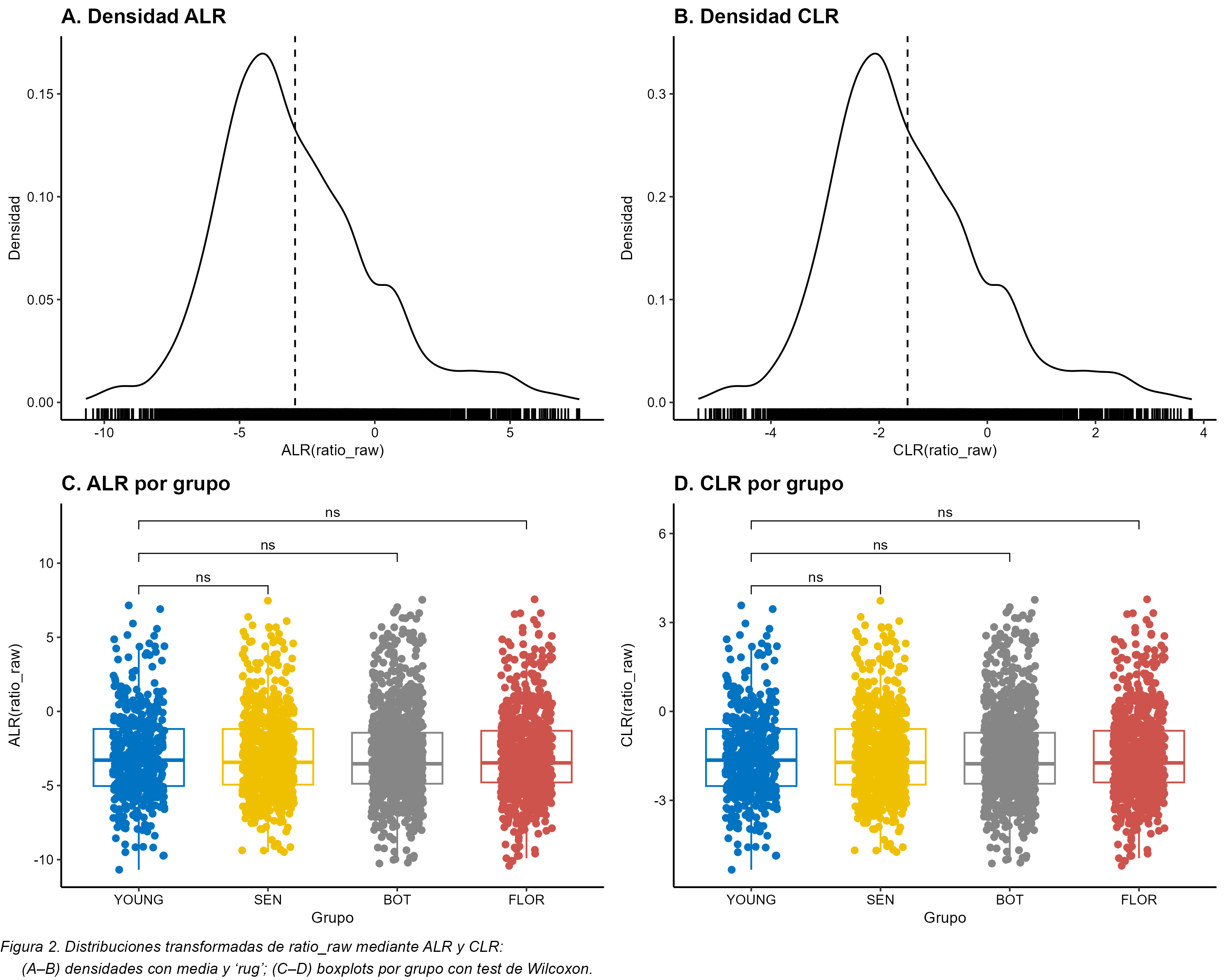
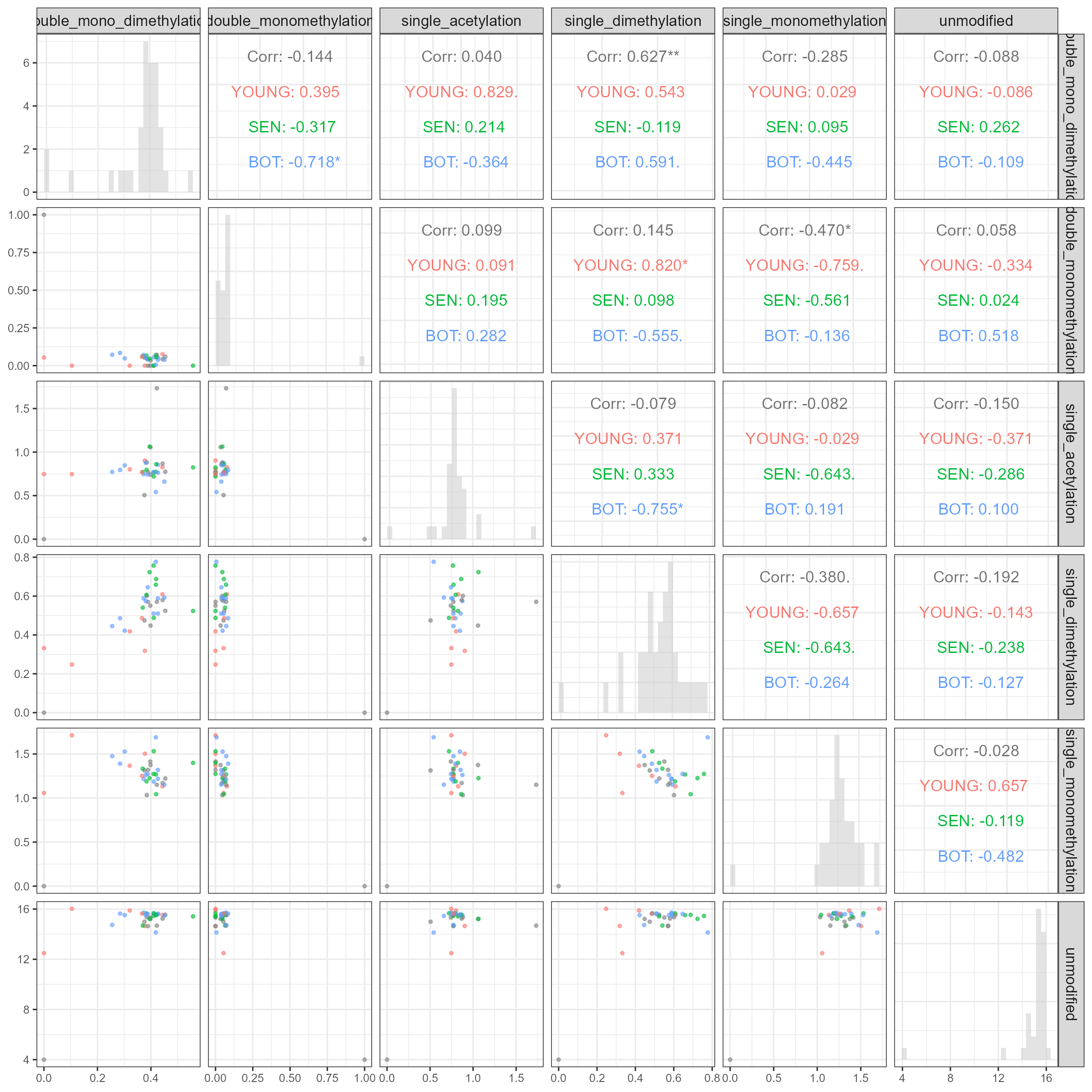
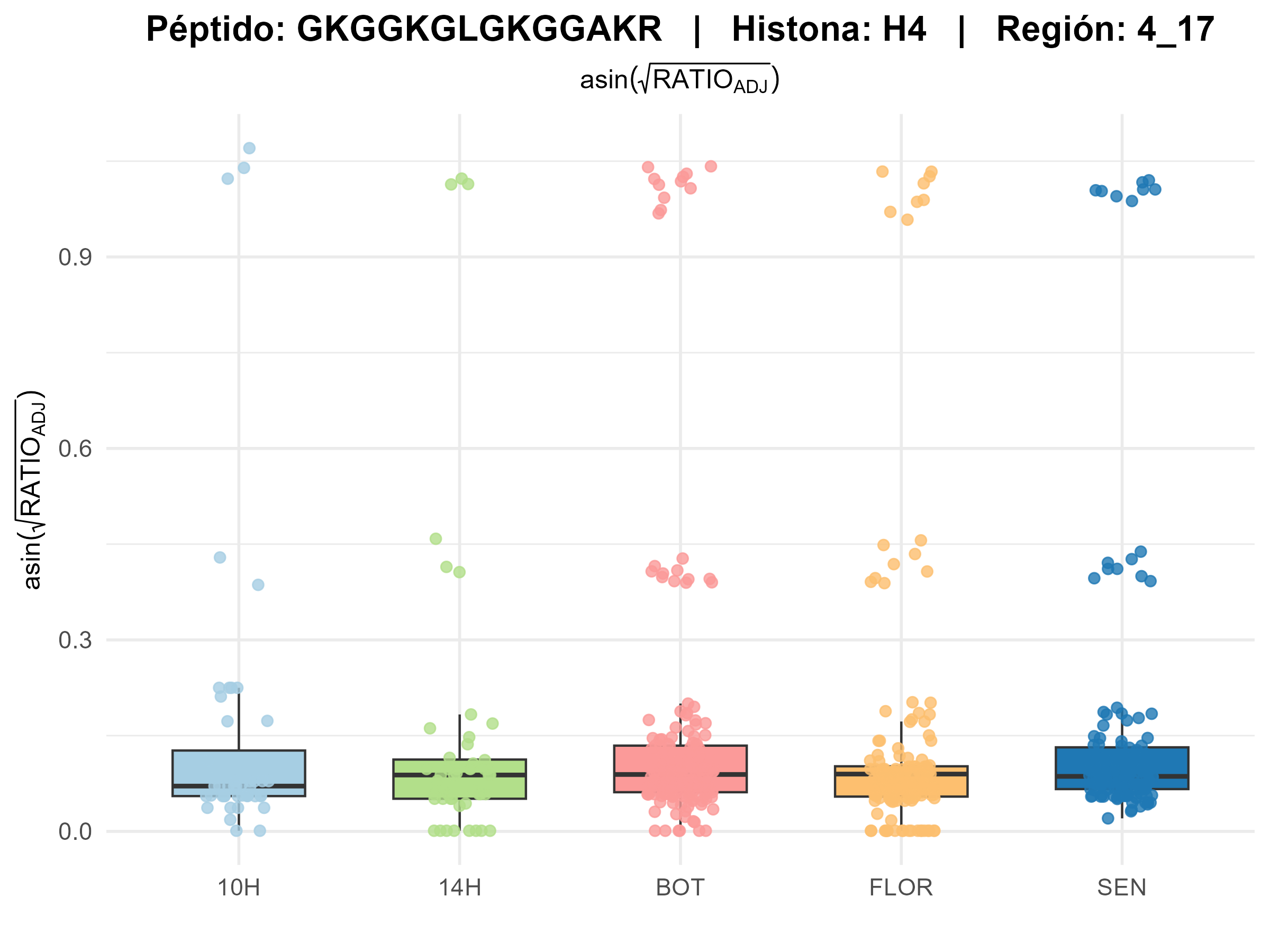
combines four chapters: (1) software validation of EpiProfile_PLANTS with
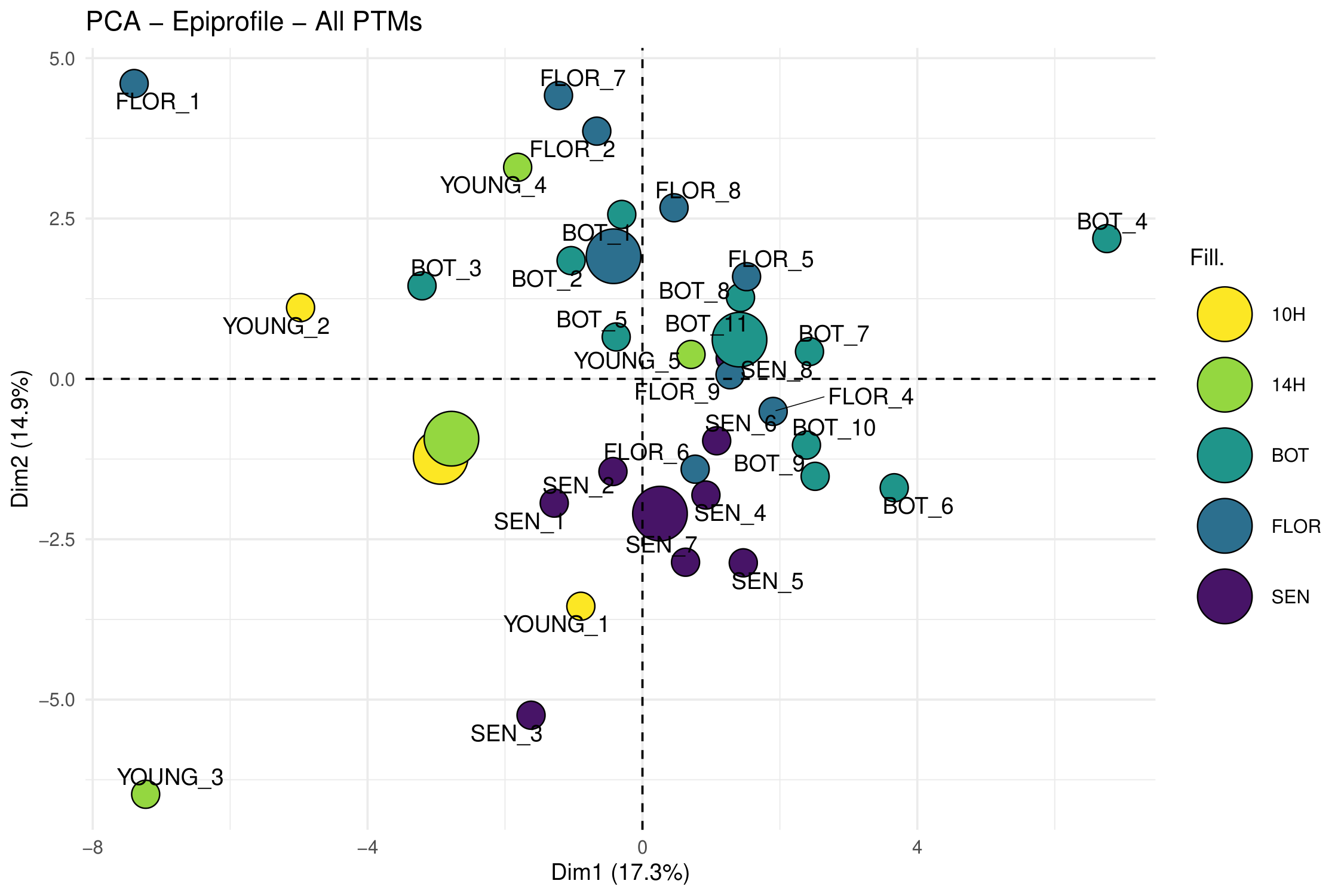
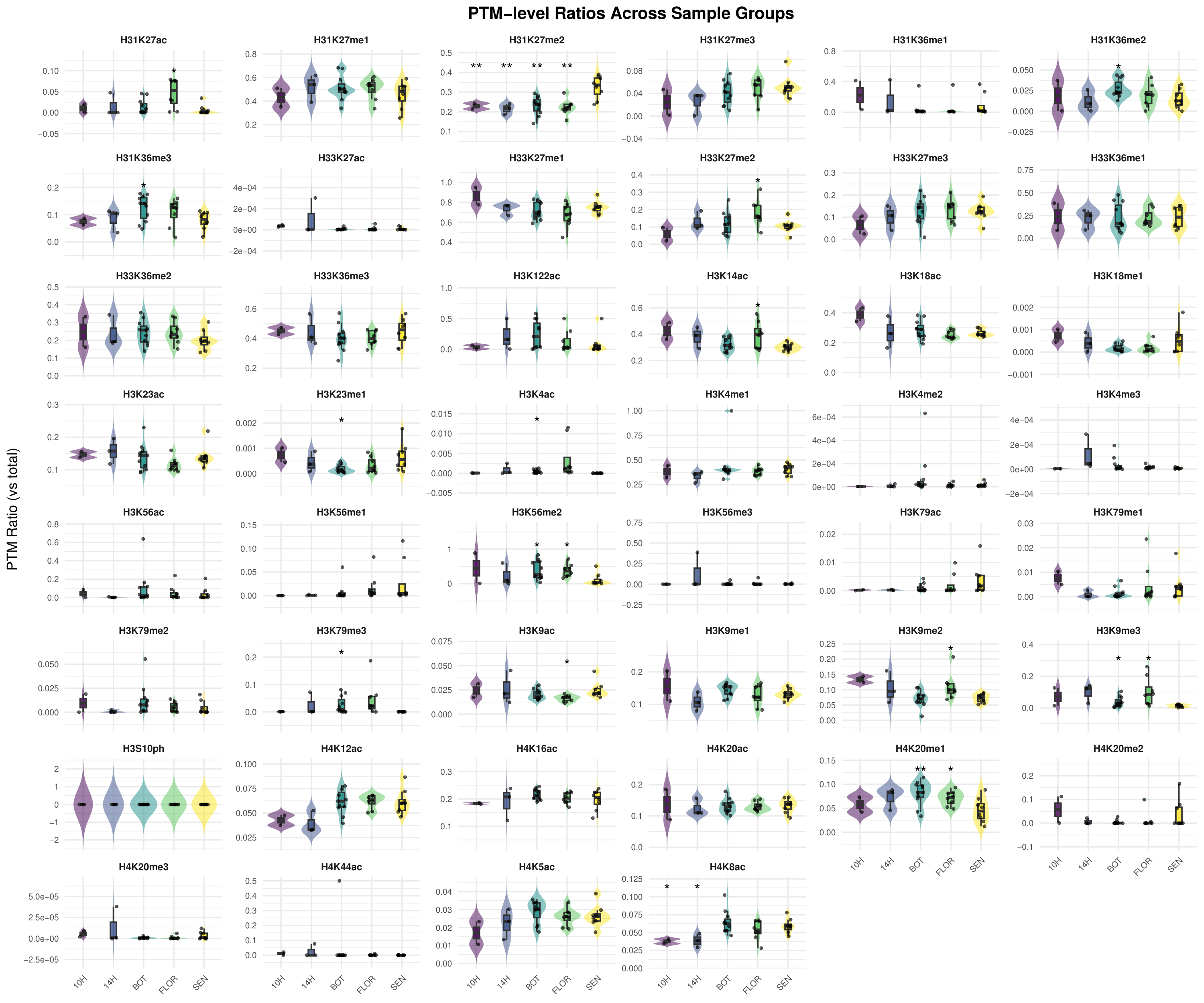
species-specific catalogs, (2) an ontogeny study of the Arabidopsis rosette across four
developmental stages (YOUNG/BOT/FLOR/SEN), (3) re-analysis of public PRIDE datasets
related to genotoxic stress, and (4) a synthesis of H3K79 behaviour across development and
stress-related contexts.
The EpiProfile_PLANTS Ecosystem
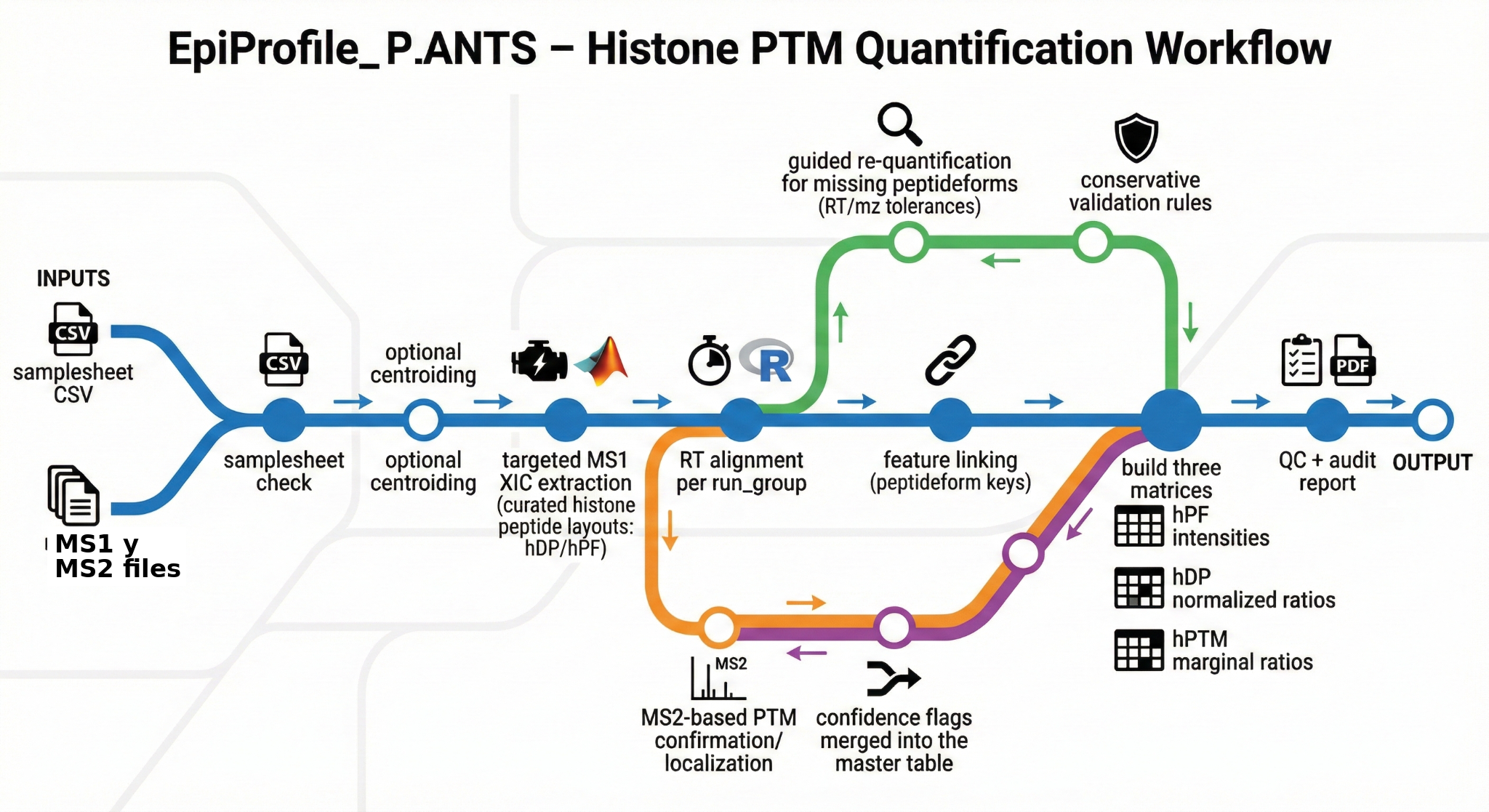
A central piece of my work is EpiProfile_PLANTS—an extension of
EpiProfile 2.0 tailored to plant histone proteomics. The ecosystem spans three interconnected
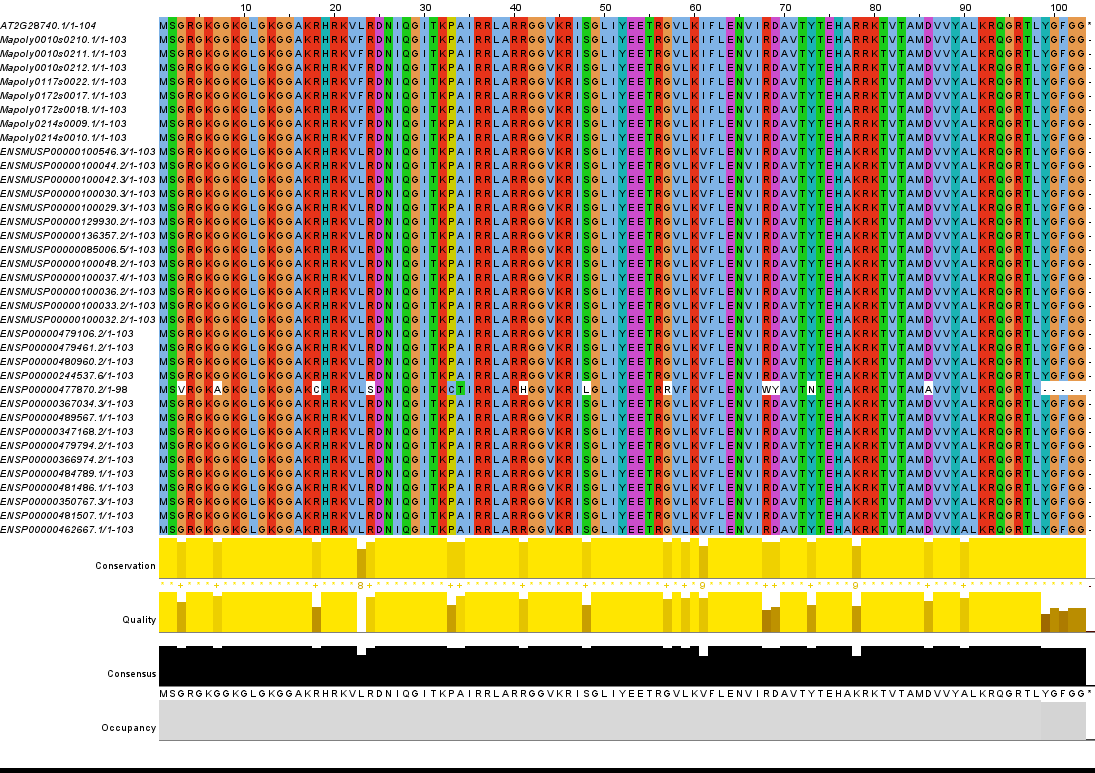
repositories: the core MATLAB code with curated peptide catalogs for
Arabidopsis, Marchantia polymorpha, and Chlamydomonas reinhardtii;
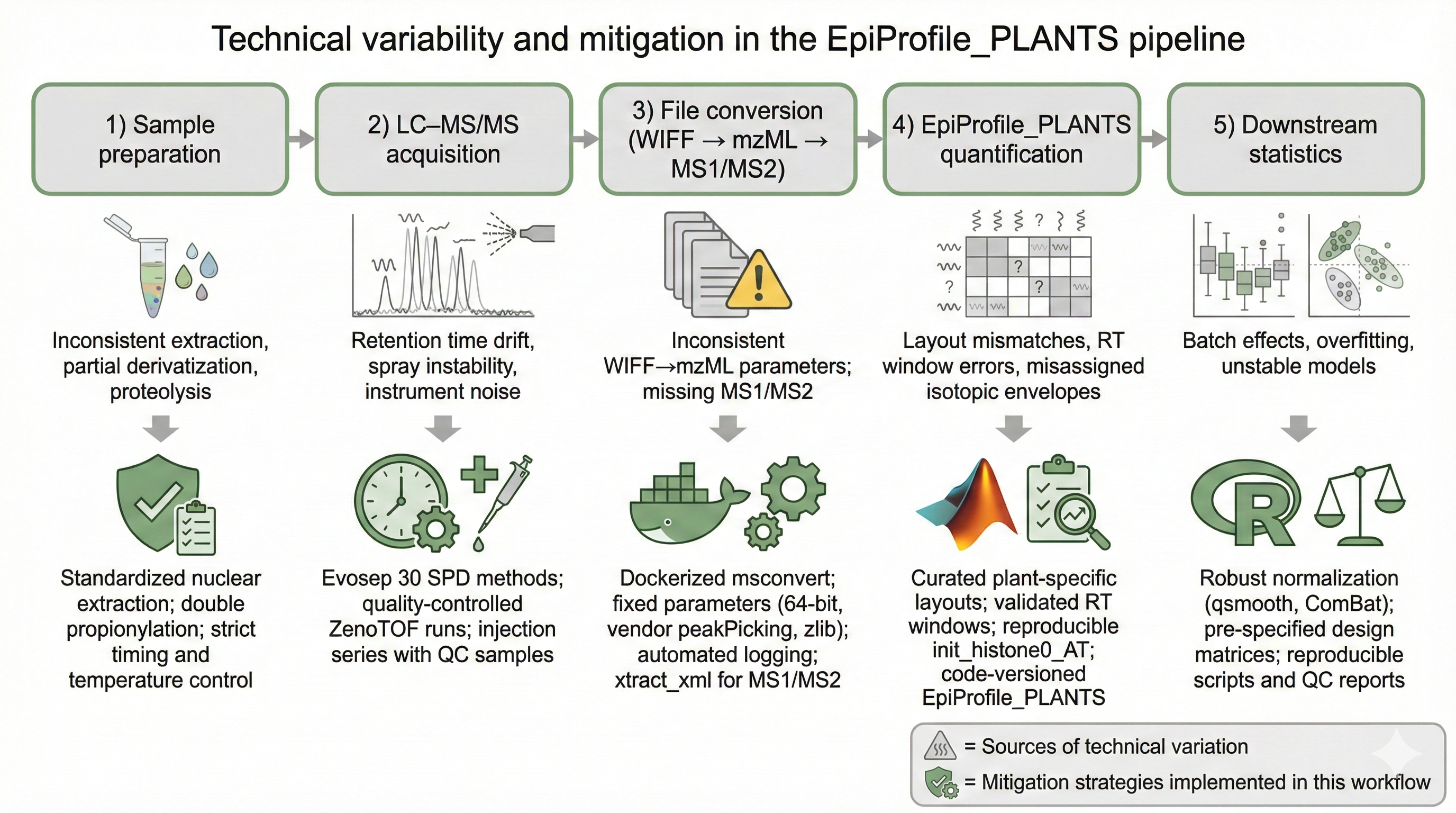
a Docker + Snakemake workflow that handles PRIDE FTP download, msconvert,
and MS1/MS2 extraction (220 raw files / 123 GB processed across PXD046034, PXD046788,
PXD014739); and an interactive Dash/Plotly dashboard with 7 analysis tabs
(heatmaps, PCA, volcano plots, PSM explorer, mass accuracy QC).
The data flows through a three-tier model: hDP (derivatized peptides) →
hPF (peptideforms) → hPTM (site-level), with T1–T4
provenance tracking and a retention-time reference system for cross-run consistency.
K-CHOPORE: Nanopore Epitranscriptomics
Beyond proteomics, I develop K-CHOPORE—a 9-stage Snakemake + Docker
pipeline for Oxford Nanopore direct RNA sequencing. It covers basecalling
(Dorado/Guppy), isoform analysis (FLAIR/StringTie2), epitranscriptomic modification detection
(ELIGOS2, m6Anet, xPore), and differential expression (DESeq2). Applied to a 2×2
factorial experiment in Arabidopsis (WT vs anac017-1), it yielded
20,958 isoforms and hundreds of DEGs. Version 3.0 adds five
non-coding RNA modules including lncRNA discovery (FEELnc, CPC2), small RNA analysis
(ShortStack, miRDeep-P2), and WGCNA co-expression networks.
Cancer Bioinformatics (CNIO)
Since November 2020 I have also been affiliated with the Computational Cancer Genomics
Group at the Centro Nacional de Investigaciones Oncológicas (CNIO, Madrid).
This experience spans NGS transcriptomics, lncRNA pipelines (VEp, FEELnc), and early-stage
cancer genomics work, and shaped how I think about scalable, auditable bioinformatics. My 2017
publication on lncRNA clusterization in head and neck squamous carcinomas came from this line of
research.
Biomedical Image Analysis (VIDIO)
VIDIO (Vision-Integrated Diagnostic Imaging Orchestrator) is my multi-modal
biomedical imaging platform, supporting retinal imaging (fundus, OCT), histopathology (OpenSlide
whole-slide images), radiology (DICOM/NIfTI), and spatial transcriptomics (10x Visium, MERFISH).
Built on Falcon WSGI, PyTorch/MONAI, OpenCV, with a PostgreSQL backend (16 tables) and TCGA/GDC
integration.
Teaching & Código Biológico
I have designed and delivered courses at the Instituto Asturiano de Administración
Pública (IAAP), the University of Oviedo, the City Council
of Oviedo, FORMACAL, and ARTEAULA, covering
Linux/WSL2/Docker, introductory Python and R/Bioconductor for omics, and hands-on projects
linking code to real biological questions.
Código Biológico is my growing outreach initiative to teach bioinformatics
from scratch—step-by-step notebooks, recorded sessions, and reusable templates
with real datasets, not toy examples.
Other Experience
Before and alongside the PhD, I have worked across multiple domains: GeoAI
(geospatial / AI-driven analysis), ICM Lugo (research and healthcare context),
and FSP-linked initiatives in regional public health. These experiences
reinforced my philosophy: workflows should be practical, documented, and teachable.
How I Work
Every analysis links back to explicit PXD accessions. Conversion from vendor
files is containerised and deterministic. Intermediates (mzML, MS1/MS2) are standardised.
Outputs are three-tier, audit-ready matrices. Documentation is citable. Code is open under
GPL-family licenses. I follow FAIR principles and believe
that if a result can’t be reproduced from raw data in a single command, it isn’t
finished yet.
Plant epiproteomics
Cancer bioinformatics
Epitranscriptomics
Biomedical imaging
FAIR pipelines
H3K79
Teaching